

Department of Physics and Astronomy: Publications and Other Research
Kirill Belashchenko Publications
Questaal: A package of electronic structure methods based on the linear muffin-tin orbital technique
Document Type
Article
Date of this Version
4-1-2020
Citation
Computer Physics Communications 249 (2020) 107065
https:// doi.org/10.1016/j.cpc.2019.107065
Abstract
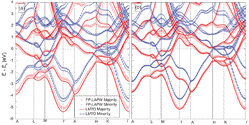
This paper summarises the theory and functionality behind Questaal, an open-source suite of codes for calculating the electronic structure and related properties of materials from first principles. The formalism of the linearised muffin-tin orbital (LMTO) method is revisited in detail and developed further by the introduction of short-ranged tight-binding basis functions for full-potential calculations. The LMTO method is presented in both Green's function and wave function formulations for bulk and layered systems. The suite's full-potential LMTO code uses a sophisticated basis and augmentation method that allows an efficient and precise solution to the band problem at different levels of theory, most importantly density functional theory, LDA+U, quasi-particle self-consistent GW and combinations of these with dynamical mean field theory. This paper details the technical and theoretical bases of these methods, their implementation in Questaal, and provides an overview of the code's design and capabilities.
Program summary: Program Title: Questaal
Program Files doi: http://dx.doi.org/10.17632/35jxxtzpdn.1
Code Ocean Capsule: https://doi.org/10.24433/CO.3778701.v1
Licensing provisions: GNU General Public License, version 3
Programming language: Fortran, C, Python, Shell Nature of problem: Highly accurate ab initio calculation of the electronic structure of periodic solids and of the resulting physical, spectroscopic and magnetic properties for diverse material classes with different strengths and kinds of electronic correlation. Solution method: The many electron problem is considered at different levels of theory: density functional theory, many body perturbation theory in the GW approximation with different degrees of self consistency (notably quasiparticle self-consistent GW) and dynamical mean field theory. The solution to the single-particle band problem is achieved in the framework of an extension to the linear muffin-tin orbital (LMTO) technique including a highly precise and efficient full-potential implementation. An advanced fully-relativistic, non-collinear implementation based on the atomic sphere approximation is used for calculating transport and magnetic properties.
Included in
Atomic, Molecular and Optical Physics Commons, Condensed Matter Physics Commons, Engineering Physics Commons, Other Materials Science and Engineering Commons, Statistical, Nonlinear, and Soft Matter Physics Commons
Comments
This is an open access article under the CC BY license