

Department of Physics and Astronomy: Publications and Other Research
Peter Dowben Publications
Document Type
Article
Date of this Version
3-29-2007
Abstract
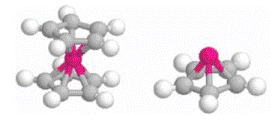
The authors report on a study of dipole flip-flop “local” transition in ferroelectric polyvinylidene fluoride [P(VDF)] chains, using total energy calculation based on the density functional theory. The calculated results indicate that a simple flipping of a single electric dipole moment is energetically allowed. Furthermore, such a flipping involves no change either in bond length, bond angle, or the orientation of the chain. The calculations also show that on a thin film of ordered chains, strong dipole interactions existing in P(VDF) could cause modulation of the dipole orientation thus forming superlattices on P(VDF) films. These results are in good agreement with recent scanning tunnel microscope experimental measurements. Furthermore, our calculations show that partial flipping may also exist and extend over a length of several monomers during the flip-flop transition.
Comments
Published by American Institute of Physics. J. Chem. Physics 126, 124908 2007. ©2007 American Institute of Physics. Permission to use. http://jcp.aip.org/.